Nicholas Abbondante, Chief EMC Engineer, Intertek Group plc03.10.16
In 2014, the International Electrotechnical Commission (IEC) published a revision of the electromagnetic compatibility (EMC) requirements for medical devices under a fourth edition of IEC 60601-1-2. The revisions included a number of changes, including robust risk analysis requirements. Under these provisions, manufacturers must submit test plans and risk analysis documents specifically focused on EMC for a new product, prior to testing. In short, the revised standard requires a thorough evaluation of EMC risk for new medical devices, and relies on documentation of that risk as a part of the approval process.
The effective date for the new standard for U.S. Food and Drug Administration (FDA) approvals is April 1, 2017, yet the agency issued a letter in 2014 recommending that devices undergo the risk management processes and EMC testing to the fourth edition standard as soon as possible. Even with more than a year until the FDA deadline is in place, however, many manufacturers may find it necessary to begin the process of EMC assessment and risk management now.
Why the Focus on EMC?
As smart, connected devices continue to take the world by storm, the medical device industry seeks to integrate the latest technology into its equipment. In turn, it also makes sense that the industry is looking to new standards as it seeks to mitigate risk and ensure compliance with EMC requirements. The revised standard looks to help mitigate risks in a constantly growing arena.
The problems that EMC could create in medical devices can range from a mild nuisance, such as an alarm going off unnecessarily, to potentially drastic effects due to device malfunction. Some examples could include:
Requirements for the Risk Management File
There are four major concepts to consider in the risk management process and, ultimately, the RMF.
Test levels should be adjusted in the RMF file if the manufacturer knows from experience, published data or representative measurements that the intended use environment has unique characteristics that would alter EM disturbance levels. If any determinations or adjustments are made, the following must be documented in the test plan and the RMF: justification for any special environments identified or adjustments made; the adjusted reasonably foreseeable maximum EM disturbance level; and the resulting final immunity test levels and details on the methods and data sources used to identify those immunity levels. Additionally, if the manufacturer is justifying a lower immunity test level in the RMF, they must also include documentation explaining why the reduced test levels are representative of the intended environment, and how it can be reasonably expected that any mitigations that might justify reduced test levels will continue to be effective over the expected service life in all locations where the equipment or system is expected to be used.
Additional requirements for the RMF include considerations for emissions testing and immunity testing, which should be documented in the test plan and report. Notes about product configuration for testing are also required and should be consistent with intended use. Product configuration information should also include all cables, tubing, containers, circuits, and special hardware or software that will be needed when the product is used as intended. It is important to be aware that determinations made in the RMF regarding the applicability of testing, product essential performance, intended environment and restrictions, and any potential loss of function the user might experience due to degradations of operation that occur during testing must eventually be documented in the information to the user, and can have a significant impact on marketing of the product.
RMF Checklist
It order to fulfill the RMF requirements, several documents of varying degrees of complexity will be required. This checklist highlights the requirements needed in an RMF which addresses EMC.
Guidelines
With the addition of the RMF requirements for EMC safety, medical device manufacturers could easily feel a little overwhelmed with the new requirements. However, with the FDA pressing for the standard to be adopted sooner than required, it’s a good idea to begin including the EMC risk management process and file for new devices. Hence, some tips to help make the process go smoothly:
Nicholas Abbondante is chief EMC engineer at Intertek. In his 13 years with the company, he has been involved in testing a wide range of radio and electronic equipment to EMC requirements for regulatory domains around the world, specializing in transmitters. Nick is a member of the TCB Council and participates in the ANSI C63.10 and ANSI C63.26 radio standards writing committees. He has a bachelor’s degree in physics from the Worcester Polytechnic Institute.
The effective date for the new standard for U.S. Food and Drug Administration (FDA) approvals is April 1, 2017, yet the agency issued a letter in 2014 recommending that devices undergo the risk management processes and EMC testing to the fourth edition standard as soon as possible. Even with more than a year until the FDA deadline is in place, however, many manufacturers may find it necessary to begin the process of EMC assessment and risk management now.
Why the Focus on EMC?
As smart, connected devices continue to take the world by storm, the medical device industry seeks to integrate the latest technology into its equipment. In turn, it also makes sense that the industry is looking to new standards as it seeks to mitigate risk and ensure compliance with EMC requirements. The revised standard looks to help mitigate risks in a constantly growing arena.
The problems that EMC could create in medical devices can range from a mild nuisance, such as an alarm going off unnecessarily, to potentially drastic effects due to device malfunction. Some examples could include:
- Issues during surgery, such as surgical table actuation, or changes in electrocardiogram (ECG) or blood pressure readings;
- Unexpected defibrillator activation
- Changing infusion rate on a pump
- Ventilator stoppage;
- Incorrect temperature, blood pressure or ECG readings;
- Reprogramming of a pacemaker or similar device; and
- Incorrect, or even useless information.
Requirements for the Risk Management File
There are four major concepts to consider in the risk management process and, ultimately, the RMF.
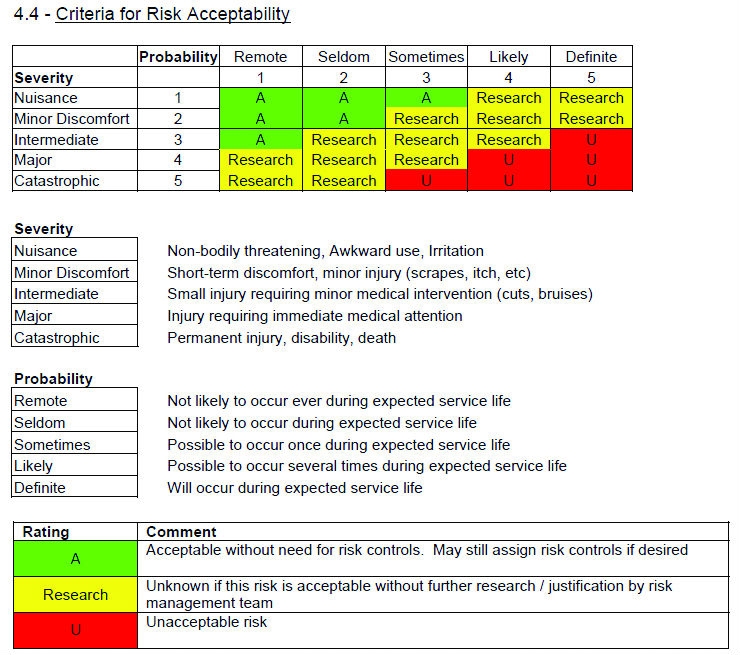
- The risk management process must comply with ISO 14971. This means that a standard operating procedure/work instruction/method that complies with the standard must be in place. The RMF will be reviewed against the requirements of this standard and all procedures must be in place.
- The RMF should clearly illustrate the process used to identify risks, as well as verify that the final document meets both the manufacturer’s needs and the minimum requirements under ISO 14971. The risk procedure outlines all of the necessary steps for creating an RMF. This might include requirements for a risk plan, hazard analysis, verification and validation procedures, or information collection during and after production. Where the procedure indicates that a step should be taken or a document generated, the manufacturer must show that those steps were followed and the risk file has been generated.
- Information related to risk management must be included in the file, as required by the ISO standard. Throughout the 60601-1 standard, pieces of information or statements are required to be documented in the RMF. Manufacturers will need to prove that the required pieces of information exist or the necessary statements are made. These must all be documented within the RMF. Fulfilling such requirements could be as easy as making a statement about expected service life, or as complicated as a structural mechanical analysis of a support system.
- Specific hazards must be evaluated through analysis and risk management procedure. The IEC 60601-1 standard lists specific hazards to be evaluated with statements such as “The hazard of XYZ shall be evaluated using the risk management process,” or “ABC shall not cause unacceptable risk as confirmed by inspection of the risk management file.” Because these hazards have been specifically identified by the IEC, the manufacturer must illustrate that the hazard/risk has been evaluated using the risk management process as required by ISO 14971. This should include identifying the hazard, determining the acceptability of the hazard creating harm, assigning risk controls for unacceptable hazards, and verifying and validating risk controls. The requirement to address EMC hazards has been explicitly outlined in the new IEC 60601-1-2 fourth edition, where it indicates that risks resulting from reasonably foreseeable electromagnetic disturbances shall be taken into account in the risk management process.
- Professional healthcare environment: Where equipment or a system will be used by healthcare professionals and is not intended for sale to the general public;
- Home healthcare: A product used in the dwelling place where a patient lives or other place where patients are present (excluding professional healthcare facilities where operators with medical training are continually available), such as domiciles, vehicles, hotels, restaurants, commercial environment, schools, and churches; and
- A special environment: An environment different from those specified above, or that require emissions limits, immunity test levels or test methods different from professional healthcare and home healthcare environments. Examples might include military areas, heavy industrial areas or a medical treatment areas with high-powered equipment.
Test levels should be adjusted in the RMF file if the manufacturer knows from experience, published data or representative measurements that the intended use environment has unique characteristics that would alter EM disturbance levels. If any determinations or adjustments are made, the following must be documented in the test plan and the RMF: justification for any special environments identified or adjustments made; the adjusted reasonably foreseeable maximum EM disturbance level; and the resulting final immunity test levels and details on the methods and data sources used to identify those immunity levels. Additionally, if the manufacturer is justifying a lower immunity test level in the RMF, they must also include documentation explaining why the reduced test levels are representative of the intended environment, and how it can be reasonably expected that any mitigations that might justify reduced test levels will continue to be effective over the expected service life in all locations where the equipment or system is expected to be used.
Additional requirements for the RMF include considerations for emissions testing and immunity testing, which should be documented in the test plan and report. Notes about product configuration for testing are also required and should be consistent with intended use. Product configuration information should also include all cables, tubing, containers, circuits, and special hardware or software that will be needed when the product is used as intended. It is important to be aware that determinations made in the RMF regarding the applicability of testing, product essential performance, intended environment and restrictions, and any potential loss of function the user might experience due to degradations of operation that occur during testing must eventually be documented in the information to the user, and can have a significant impact on marketing of the product.
RMF Checklist
It order to fulfill the RMF requirements, several documents of varying degrees of complexity will be required. This checklist highlights the requirements needed in an RMF which addresses EMC.
- Identification of intended environment and applicable standards;
- Specific details of immunity pass/fail criteria;
- Determination of how the product will be monitored to demonstrate compliance with the immunity pass/fail criteria;
- Acceptable degradations for immunity pass/fail criteria;
- Configurations for testing most likely to result in unacceptable risk;
- Modes and settings most likely to result in unacceptable risk;
- Justification for and special increased or decreased test levels
- Identification of product risk frequencies;
- If applicable, confirmation of ESG exposure and application of ESG testing;
- Evaluation of non-medical equipment used in the system, including whether it could affect basic safety or essential performance and whether it should be tested to IEC 60601-1-2 or other applicable standards;
- List of radio services the device could potentially be exposed to, with output power, frequency of operation, modulation type and expected separation distance;
- AC/DC power supply specifications;
- Indication of expected service life.
Guidelines
With the addition of the RMF requirements for EMC safety, medical device manufacturers could easily feel a little overwhelmed with the new requirements. However, with the FDA pressing for the standard to be adopted sooner than required, it’s a good idea to begin including the EMC risk management process and file for new devices. Hence, some tips to help make the process go smoothly:
- Don’t get bogged down in questions about RMF format. There is no one set format to an RMF, so instead of focusing on an elaborate presentation of the required information, just keep it simple. A basic approach is the best one to take.
- Remember the particular standards that may apply to your product. This may modify the basic performance criteria and test requirements. Along these lines, familiarize yourself with the language of 60601-1-2, so that you can identify particular requirements as they relate to your device.
- Review the RMF to ensure that it includes all of the required items and any supplemental information. You also need to make sure that the RMF addresses all reasonable risks associated with EMC.
- Check to make sure the risk evaluations makes sense and there are no obvious hazards missing in the RMF documents. Also, check to ensure the hazard weighting is reasonable for the device at hand.
- Use a measured approach when evaluating and weighting hazards. Ultimately, the manufacturer is responsible for the risk analysis and RMF, so employ due diligence in your analysis and documentation.
Nicholas Abbondante is chief EMC engineer at Intertek. In his 13 years with the company, he has been involved in testing a wide range of radio and electronic equipment to EMC requirements for regulatory domains around the world, specializing in transmitters. Nick is a member of the TCB Council and participates in the ANSI C63.10 and ANSI C63.26 radio standards writing committees. He has a bachelor’s degree in physics from the Worcester Polytechnic Institute.